After a long review process, CONSULT-II finally appeared in Bioinformatics. I think CONSULT-II is conceptually very exciting but as a software I am not very proud of it. The implementation is very eclectic and not user-friendly at all. Luckily, I was able to re-implement it for KRANK, and KRANK’s software offers the same functionality with an improved user experience and increase speed. Another thing is that, CONSULT-II consists of many heuristics that there are alternatives for. We have not explored much of these alternatives. I am not quite sure how promising to do so, investing too much time to compare different heuristics does not feel very productive. Anyway, I enjoyed working on this project as it was a nice introduction to metagenomics for me. I used C++ after many years, and (hopefully) gained some experience to use better practices in the next project. Here is the abstract:
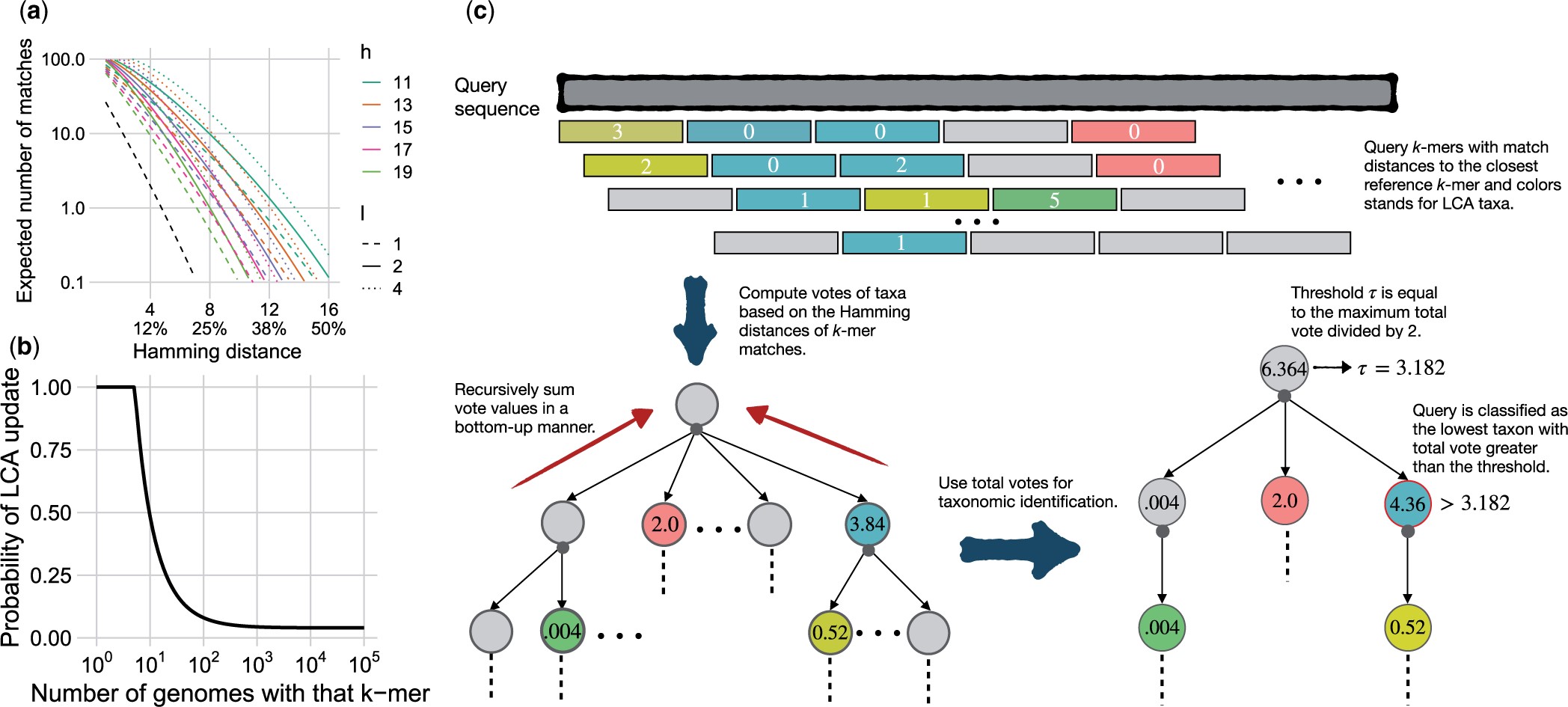
Motivation: Taxonomic classification of short reads and taxonomic profiling of metagenomic samples are well-studied yet challenging problems. The presence of species belonging to groups without close representation in a reference dataset is particularly challenging. While k-mer-based methods have performed well in terms of running time and accuracy, they tend to have reduced accuracy for such novel species. Thus, there is a growing need for methods that combine the scalability of k-mers with increased sensitivity.
Results: Here, we show that using locality-sensitive hashing (LSH) can increase the sensitivity of the k-mer-based search. Our method, which combines LSH with several heuristics techniques including soft lowest common ancestor labeling and voting, is more accurate than alternatives in both taxonomic classification of individual reads and abundance profiling.
Availability and implementation: CONSULT-II is implemented in C++, and the software, together with reference libraries, is publicly available on GitHub https://github.com/bo1929/CONSULT-II.